Le génome à la loupe : principe de la GWAS
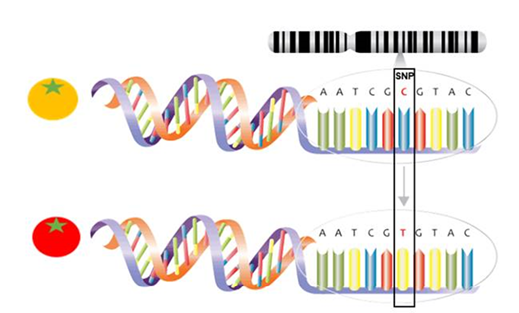
Le génome de la plupart des êtres vivants est constitué d’ADN, lui-même composé de nucléotides. On observe de nombreuses variations entre les nucléotides de deux individus : on parle de polymorphisme mononucléotidique ou SNP. Ces variations sont à l’origine de caractères différents, tels que la taille, la couleur, ou la résistance aux maladies par exemple. Mais comment déterminer les SNP qui sont corrélés à ces caractères ? C’est là qu’intervient la GWAS (Genome Wide Association Study ou étude d’association pangénomique). Utilisée depuis le milieu des années 2000, cette méthode d’analyse puissante utilise des informations de phénotypage et de génotypage sur une large population d’individus avec un grand nombre de marqueurs moléculaires afin d’identifier les SNP associés à des caractères d’intérêt.
La GWAS, quésaco ?
La GWAS (Genome Wide Association Study ou étude d’association pangénomique) consiste à identifier des variations nucléotidiques ou SNP associés à un caractère d’intérêt. Elle a d’abord été utilisée en génétique humaine au milieu des années 2000, puis en sélection animale, et elle est aujourd’hui de plus en plus utilisée dans le domaine végétal.
Un marqueur moléculaire identifie, à un endroit donné du génome, le ou les allèles présents pour un individu et permet de révéler les différences entre individus. Lorsque l’information allélique permet de prédire les variations d’un caractère, on parle d’association ou de liaison entre le marqueur et le caractère. Le caractère étudié peut aussi bien être la couleur de la fleur que la forme des feuilles, la taille des fruits ou encore la résistance ou la sensibilité à une maladie par exemple. La seule condition pour étudier un caractère est qu’il soit quantifiable et héritable, c’est-à-dire qu’il se transmette au moins partiellement via les gènes.
Utilisation des résultats d’une GWAS
L’identification de marqueurs moléculaires corrélés à un caractère d’intérêt apporte une information sur la position d’un ou plusieurs gènes contrôlant le caractère en question. L’identification de ce ou ces gènes permet de mieux comprendre les mécanismes biologiques impliqués dans l’expression du caractère.
De plus, l’identification de variants SNP liés au caractère d’intérêt peut permettre d’utiliser par la suite ces marqueurs moléculaires dans des programmes de sélection assistée par marqueurs (SAM). Le génotypage des individus à l’aide de ces marqueurs moléculaires permet alors au sélectionneur de prédire leur phénotype avant le développement complet de la plante (à partir d’un simple prélèvement de tissu foliaire non destructif). Le sélectionneur peut alors rapidement trier ses plantes et gagner du temps dans ses cycles de sélection.
Quel matériel végétal utiliser ?

La GWAS vérifie l’effet des allèles sur le caractère par des tests de corrélation entre le phénotype (le caractère) et le génotype de différents individus. Pour cela, la population d’étude doit être non apparentée et diversifiée à la fois phénotypiquement et génétiquement. Pour maximiser cette diversité, il faut croiser les ressources génétiques, regrouper différentes origines géographiques, récupérer les semences de différentes banques lorsque c’est possible, intégrer des écotypes sauvages… Dans certains cas, des populations composées d’un nombre restreint d’individus maximisant la diversité sont construites spécifiquement, il s’agit des core-collections.
Des milliers de marqueurs

Concernant le génotypage, les marqueurs moléculaires utilisés doivent permettre de distinguer les individus, c’est-à-dire ne pas être systématiquement présents dans la population d’étude, ni être associés à un seul individu. La densité de marqueurs doit être élevée pour pouvoir identifier une région restreinte associée au caractère. L’essor des nouvelles technologies de génotypage à haut débit a ouvert la voie à l’obtention rapide d’un très grand nombre de données de génotypage, notamment par l’utilisation de puces à ADN, capables de caractériser des milliers de marqueurs SNP en un temps record.
Les puces sont conçues pour une espèce donnée avec les marqueurs les plus performants répartis sur l’ensemble du génome.
Phénotypage de la population

Les régions du génome identifiées par une étude de GWAS sont appelés des QTL : Quantitatif Trait Locus (loci). Un caractère peut être contrôlé par un ou plusieurs QTL expliquant chacun une part de variation du caractère. L’analyse se base donc sur la variation du caractère, c’est pourquoi le caractère doit être noté le plus précisément possible et de manière quantitative. Plus la variable de notation sera précise, plus l’analyse sera puissante. Chaque individu de la population sera noté sur cette variable avec des conditions de culture et de notation identiques pour toute la population.
Quelques exemples de caractères et leurs variables de notation :
- Résistance à une maladie évaluée par un pourcentage de surface contaminée ;
- Sucres évalués par une concentration en ppm ;
- Développement du fruit estimé par son diamètre (ou largeur), son poids ou encore sa densité.
Association statistiques « génotype-phénotype »
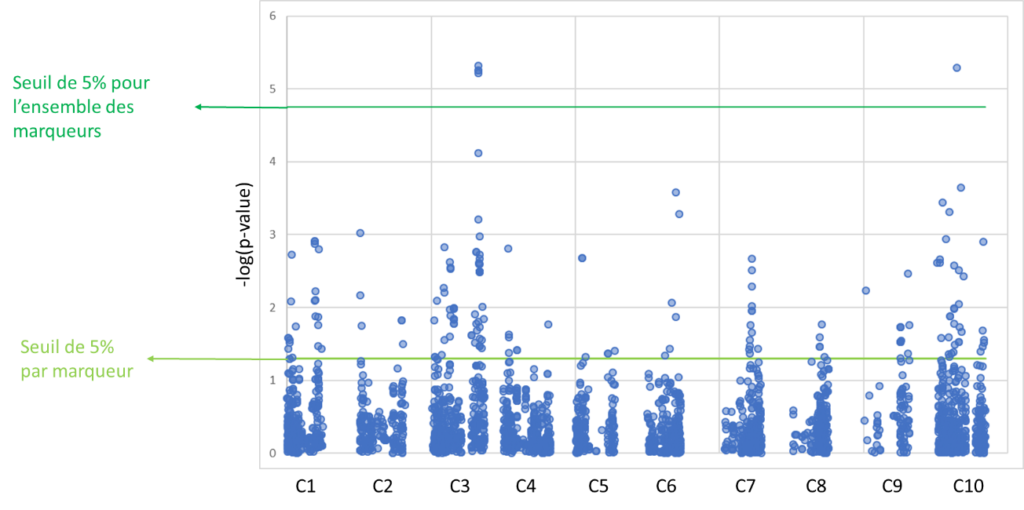
Le schéma classique des résultats d’une GWAS est le Manhattan plot : -log(p-value) pour tous les marqueurs.
Un point correspond à la significativité du test de corrélation entre un marqueur et le caractère d’intérêt. L’abscisse donne la position du marqueur et par conséquent son identité et l’ordonnée la p-value du test, ou plutôt le –log(p-value). Plus la p-value est faible plus l’effet du marqueur sur le caractère sera significatif (faible taux d’erreur pour l’association) et plus l’opposé du log sera élevé. Ainsi les régions présentant des pics sur le schéma sont des régions présentant des gènes associés au caractère étudié. Chaque bloc correspond à l’ensemble des marqueurs d’un même chromosome : de gauche à droite = de haut en bas du chromosome.
Conclusion
La génétique d’association est une approche puissante pour identifier des régions génomiques contrôlant des caractères quantitatifs d’intérêt chez les plantes et les marqueurs moléculaires associés à ces régions.
Ces marqueurs peuvent ensuite être utilisés par les sélectionneurs pour optimiser et accélérer leurs schémas de création variétale.
Sources
Bush, William S., et Jason H. Moore. « Chapter 11: Genome-Wide Association Studies ». Édité par Fran Lewitter et Maricel Kann. PLoS Computational Biology 8, no 12 (27 décembre 2012): e1002822. https://doi.org/10.1371/journal.pcbi.1002822.
Crédits photos : Vegenov
Bonsoir, ces informations sur le principe du GWAS m’ont été d’une utilité.
En effet, je suis un étudiant en thèse au Burkina Faso, travaillant sur le mung bean (VIGNA RADIATA (L.) R. WILCZEK).
Mon etude est orientée sur GWAS.
Ma question est de savoir si vous pouvez m’aider avec plus de documents sur le sujet afin d’améliorer ma connaissance sur l’etude.
Merci
Bonjour,
Merci de l’intérêt porté à notre billet. Nous n’avons pas eu l’occasion encore de travailler sur le haricot mungo à Vegenov et ne disposons pas de bibliographie sur cette espèce. Pour les recherches de publications, il vous est possible de chercher des articles sur les bases de données et moteurs de recherche scientifiques (PubMed, Google Scholar, Sciences Direct…) en associant les mots clés suivants : Mung bean, Vigna radiata, GWAS, Genome-Wide Association Analysis, SNP, Genotyping by Sequencing (GBS), Genome-Wide SNP, Association Mapping. Bien cordialement,
Bonjour, je suis AKA de la Côte d’Ivoire. Je travaille pour ma thèse sur le manioc. Ce billet m’a permis de comprendre beaucoup de choses sur le GWAS. Cependant, j’aimerais savoir pour ce type d’étude, combien de cultures multi locales je dois faire, quel dispositif serait idéal et combien de temps ou saison me faut-il pour faire une bonne étude GWAS?
Bonjour, Pour plus d’information, vous pouvez contacter Charlotte Roby, chargée de projets Vegenov en Amélioration et Traçabilité des Plantes (roby@vegenov.com). Bien cordialement,